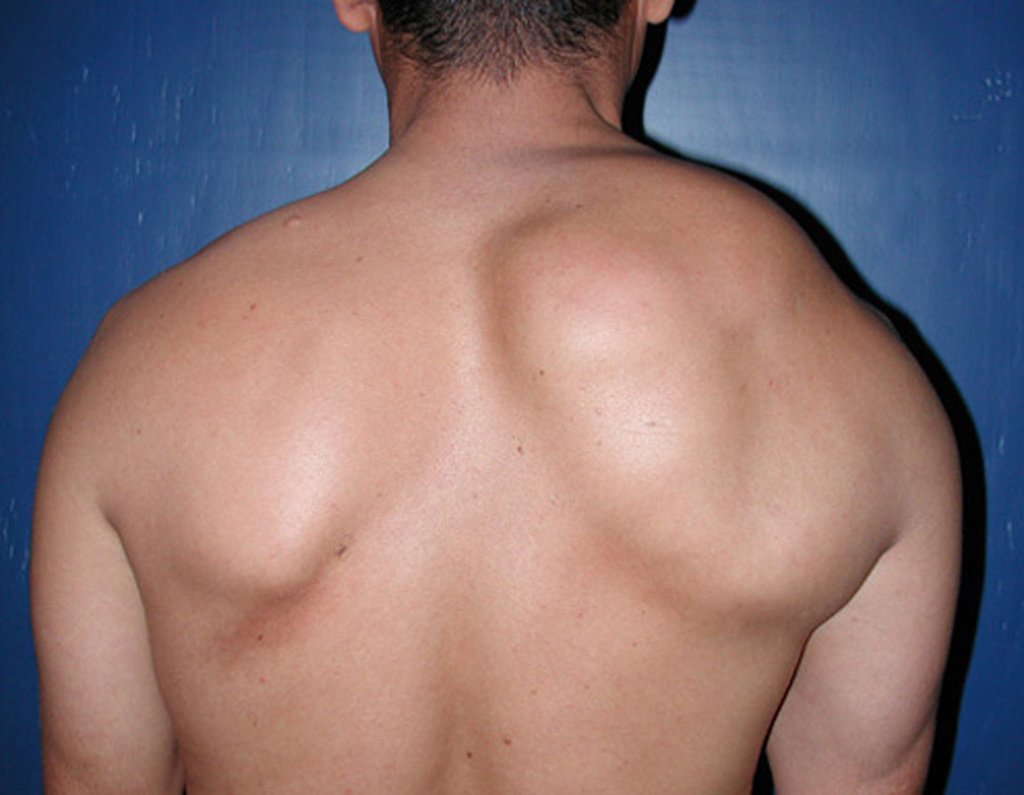
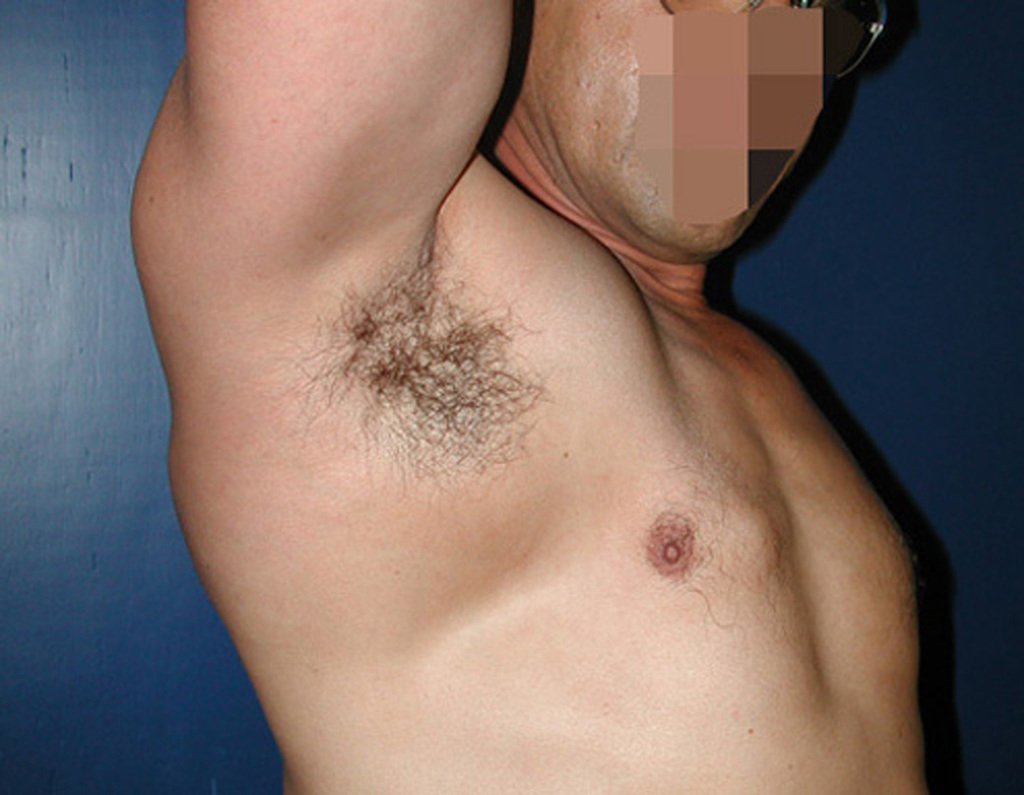
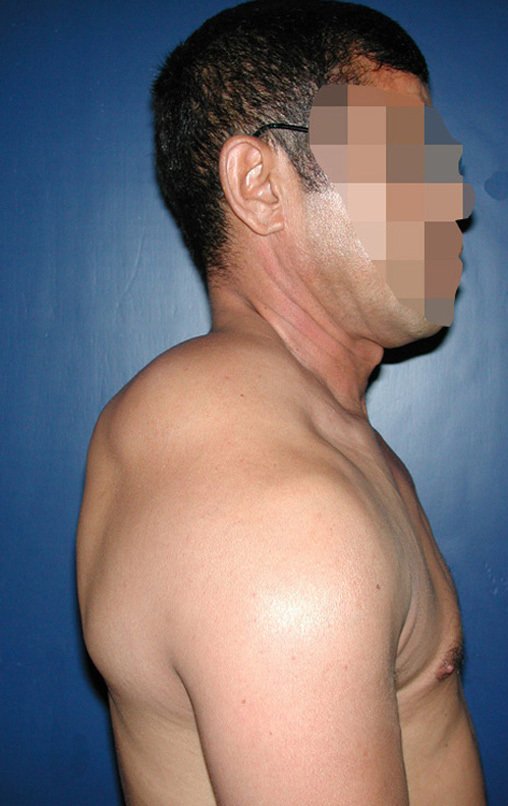
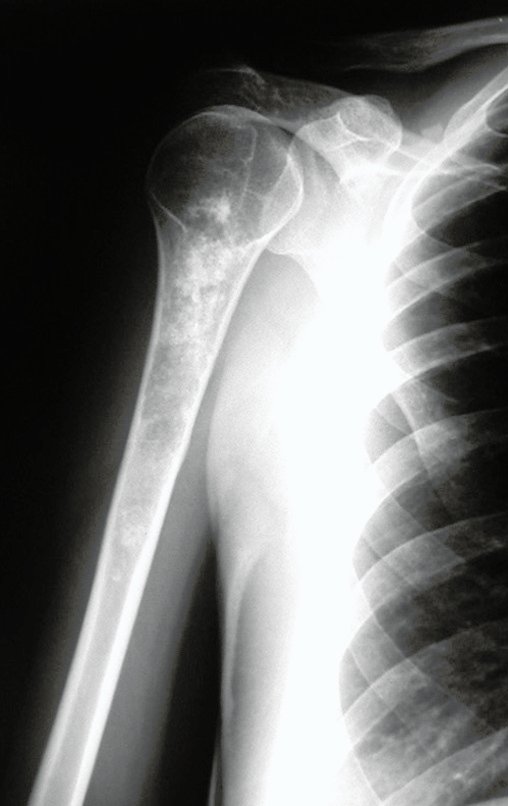
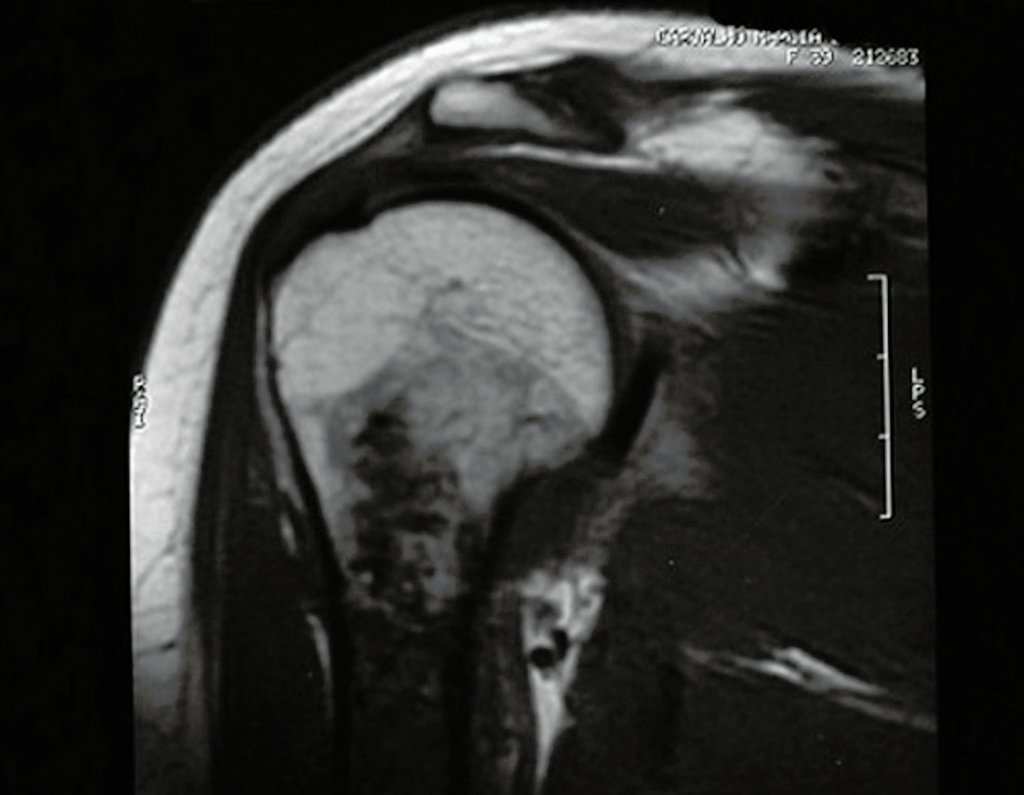
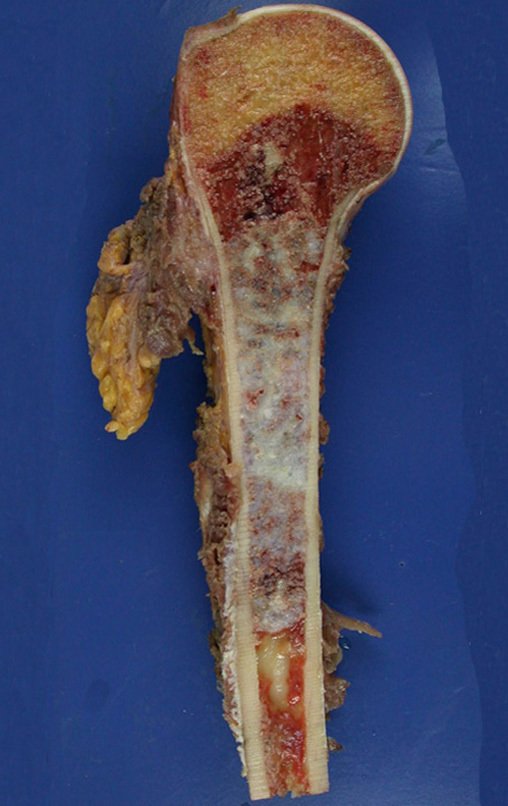
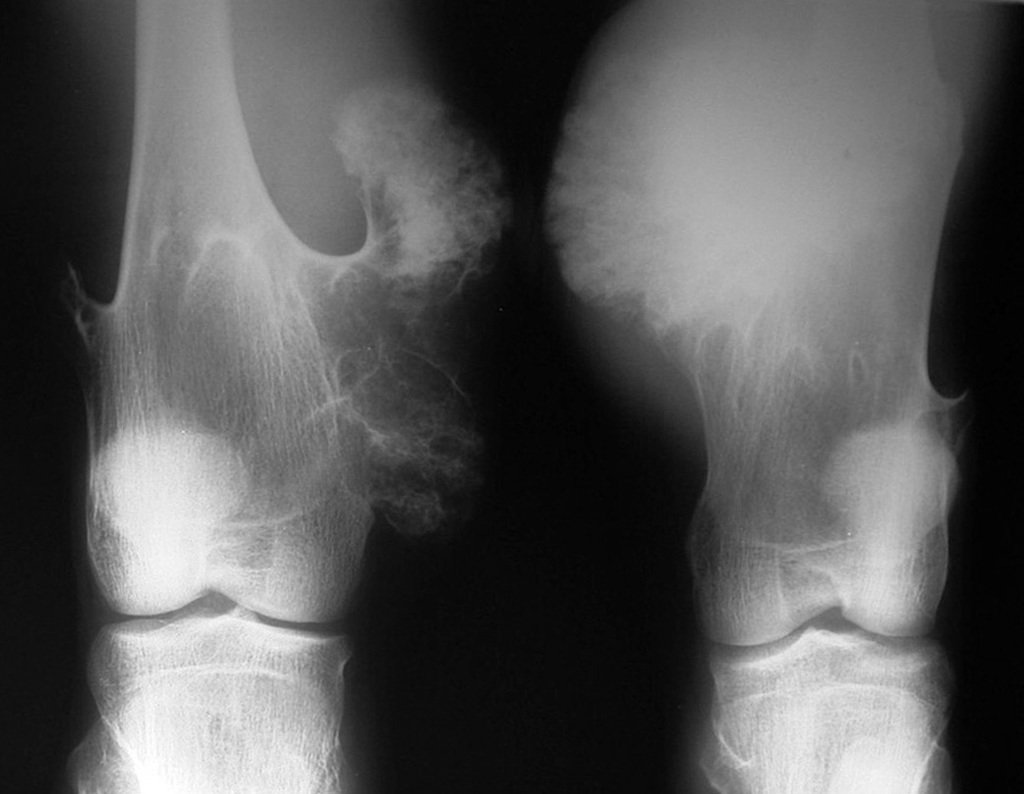
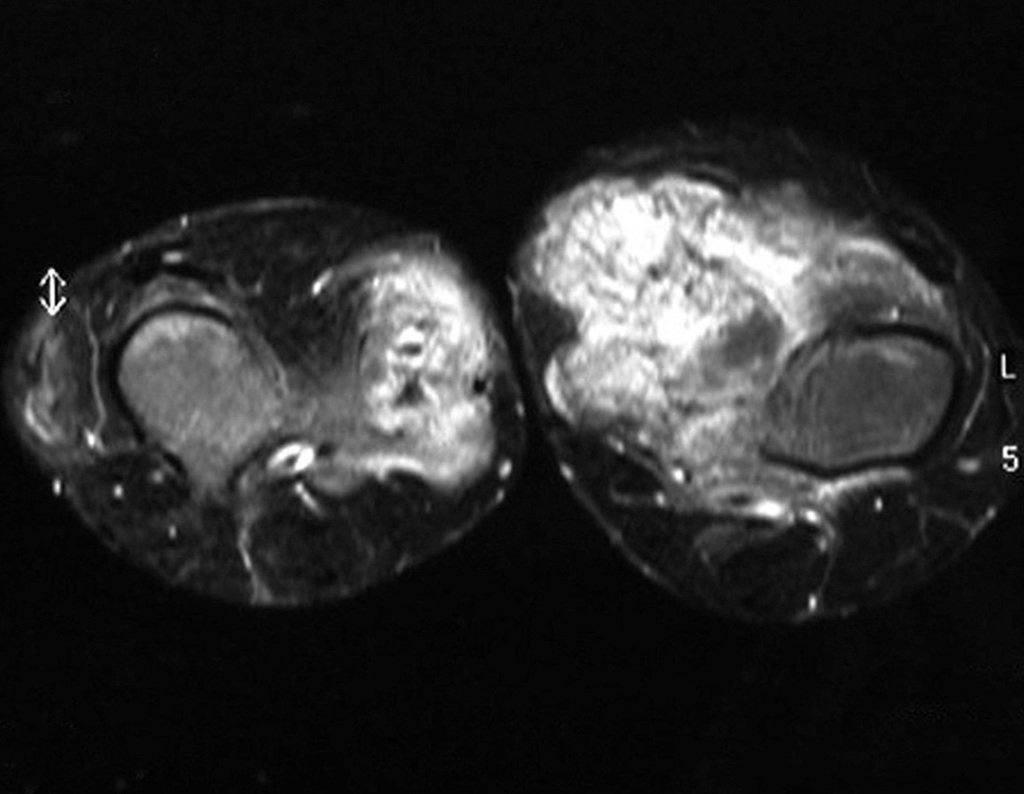
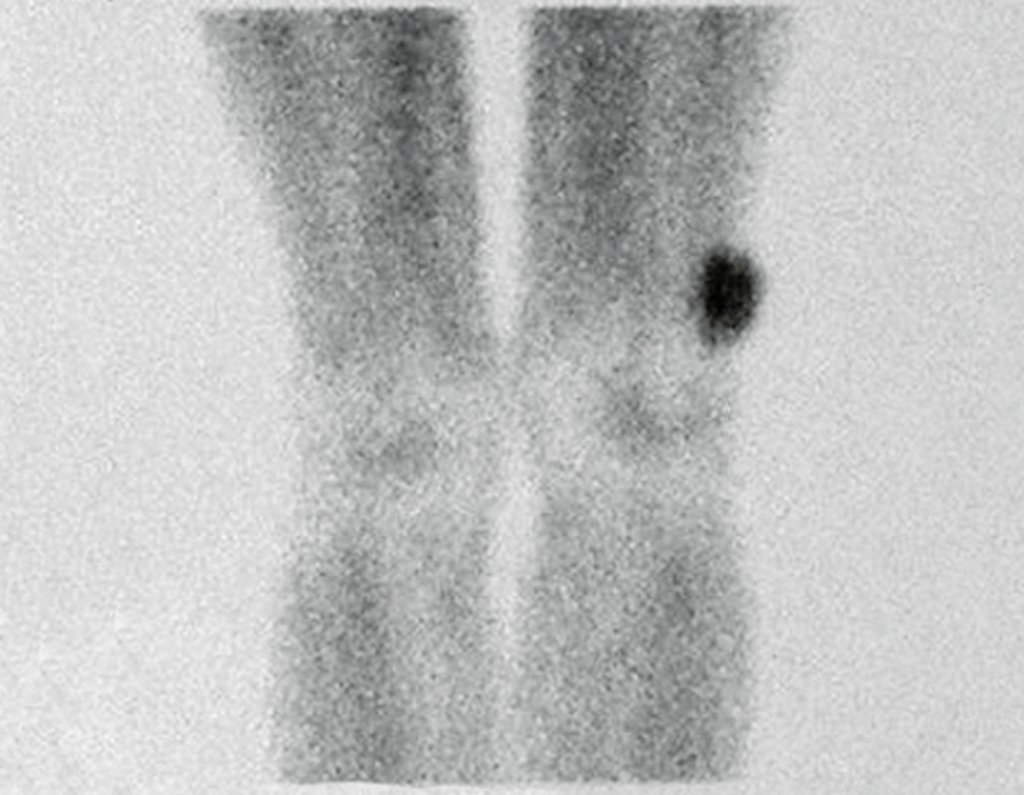
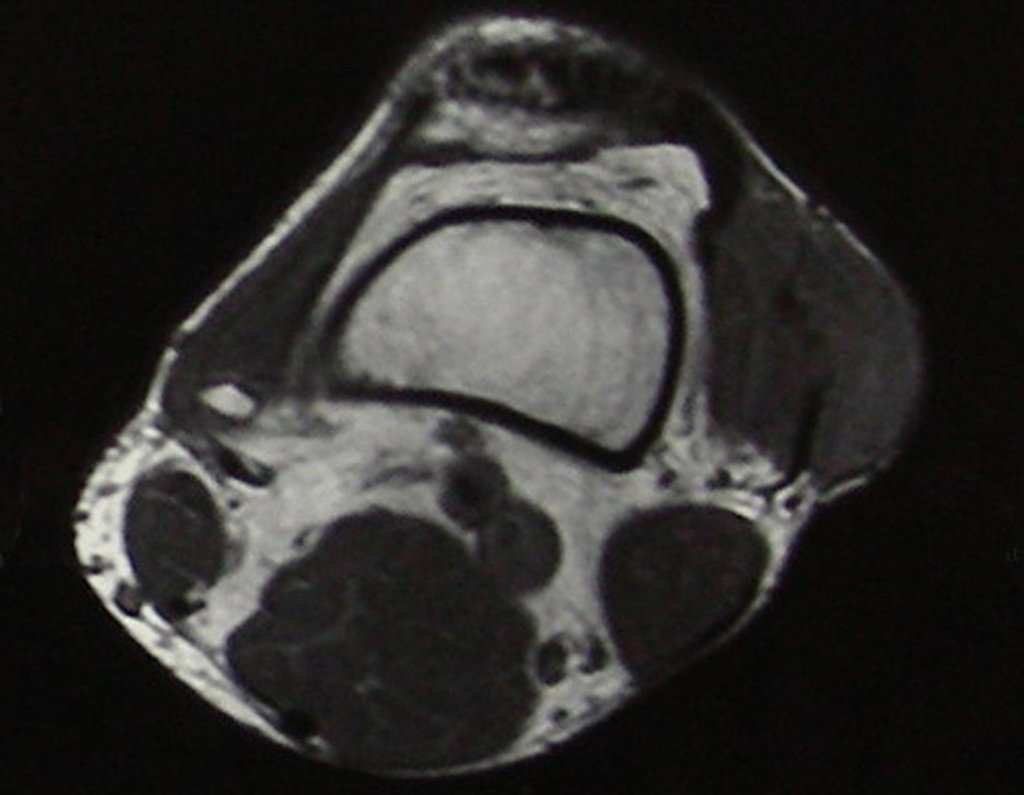
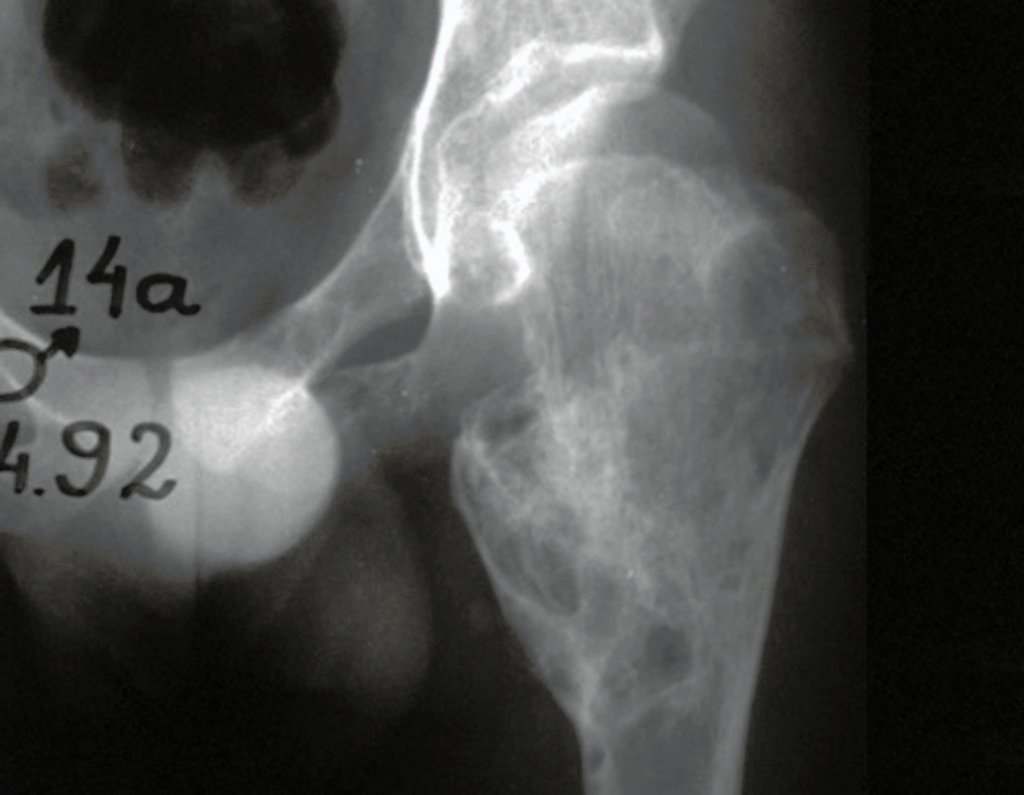
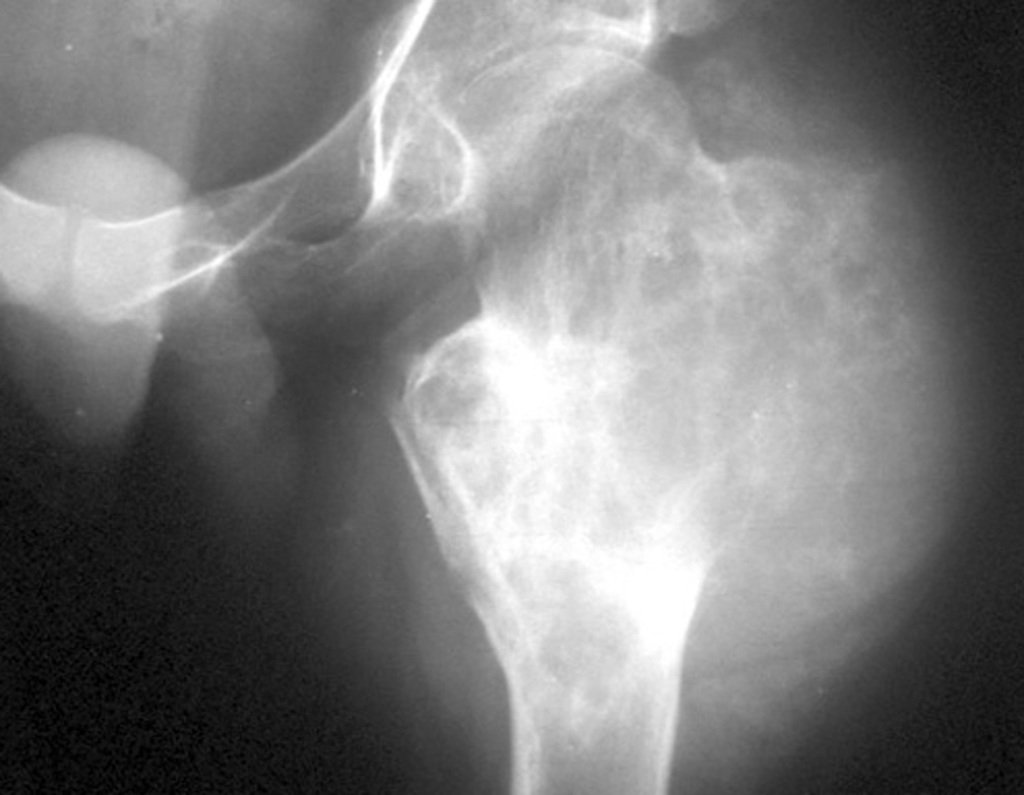
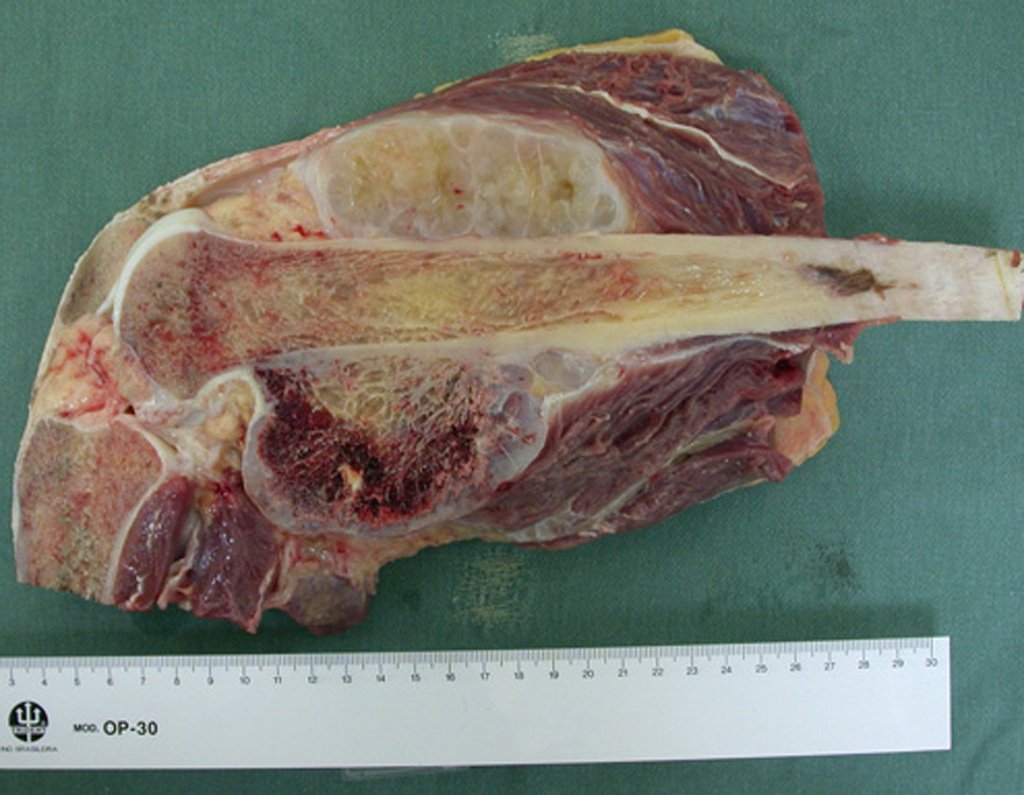
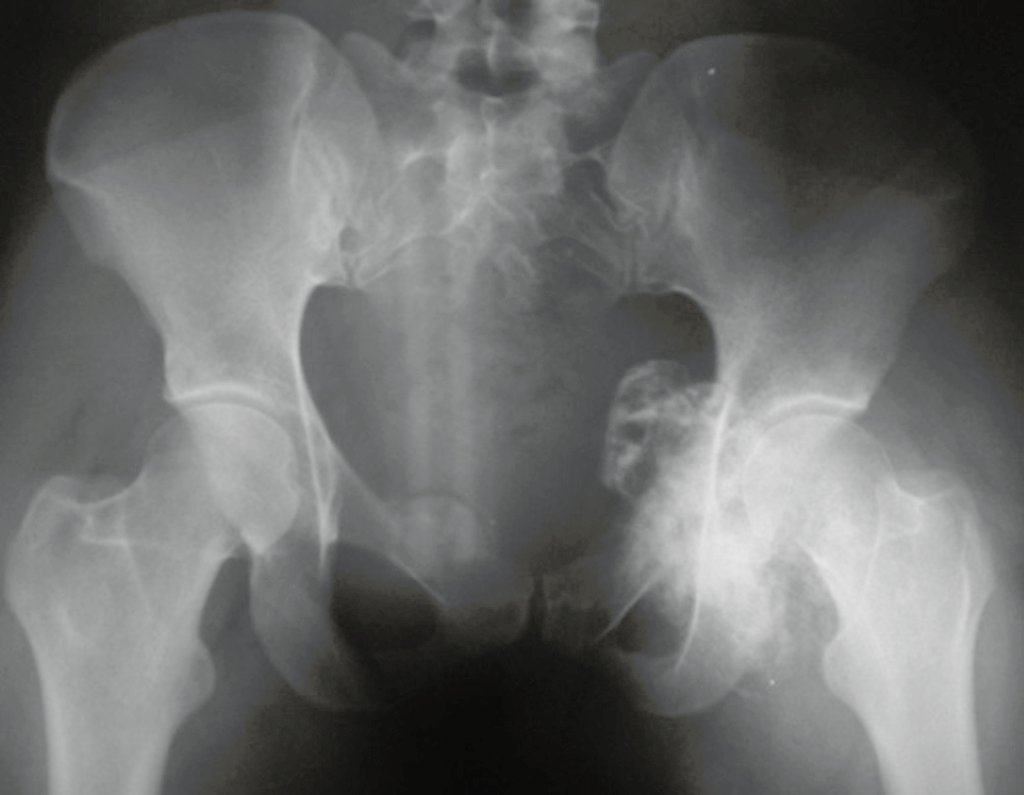
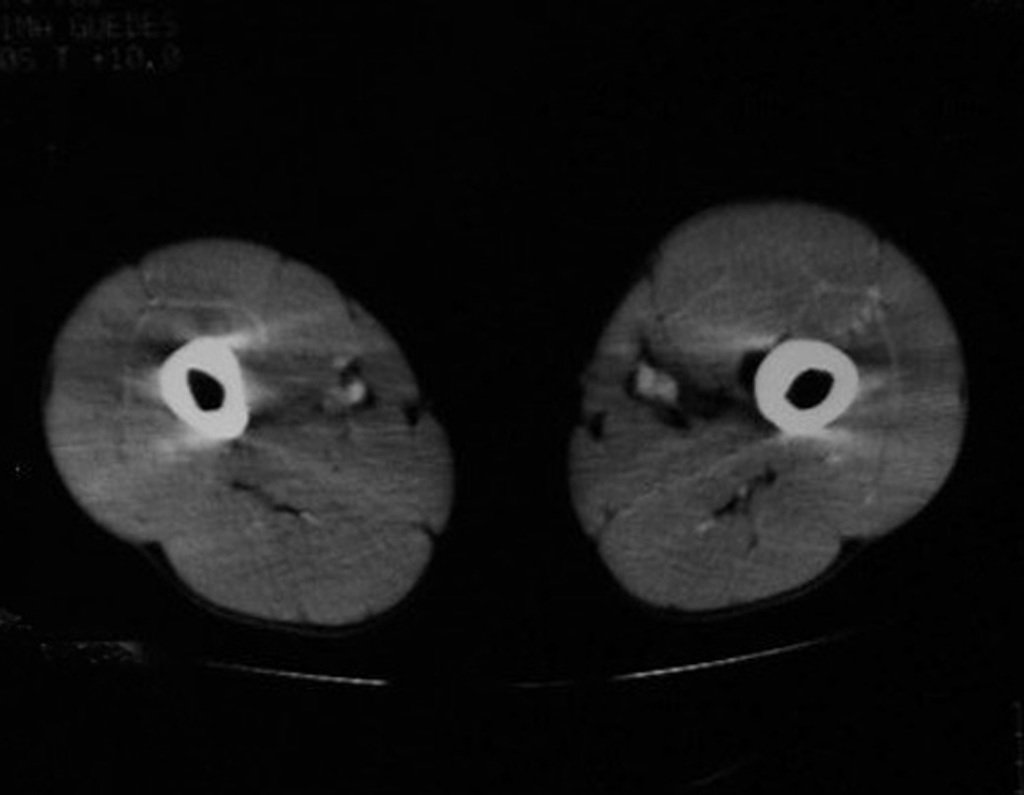
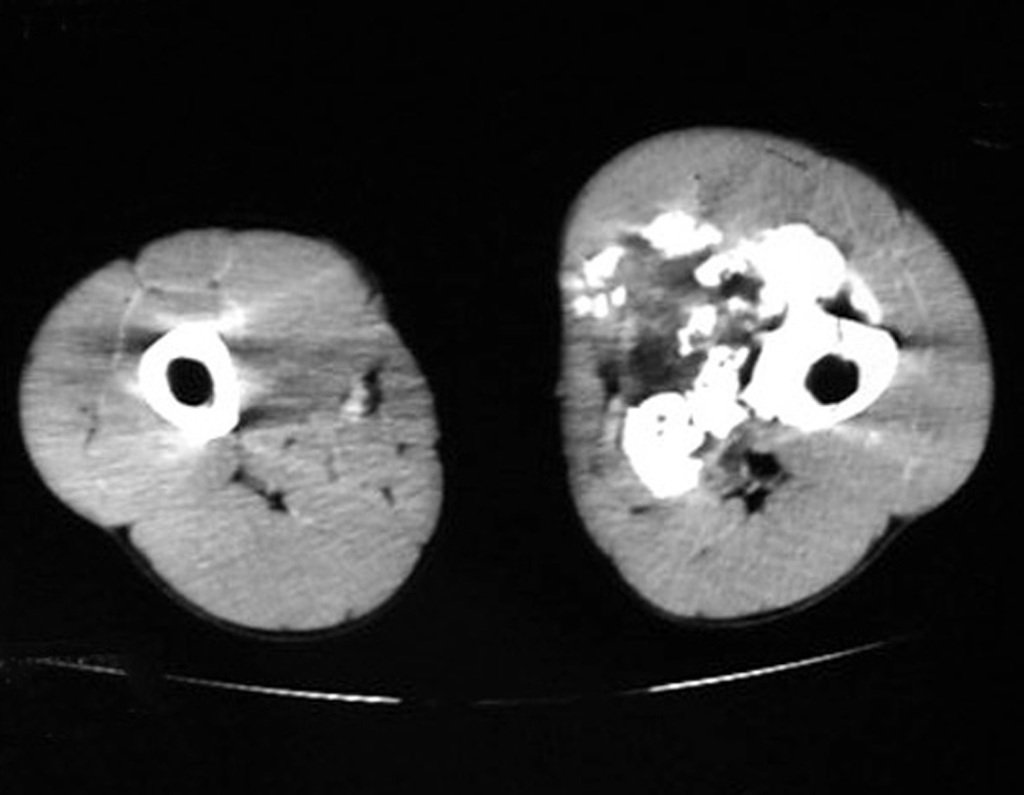
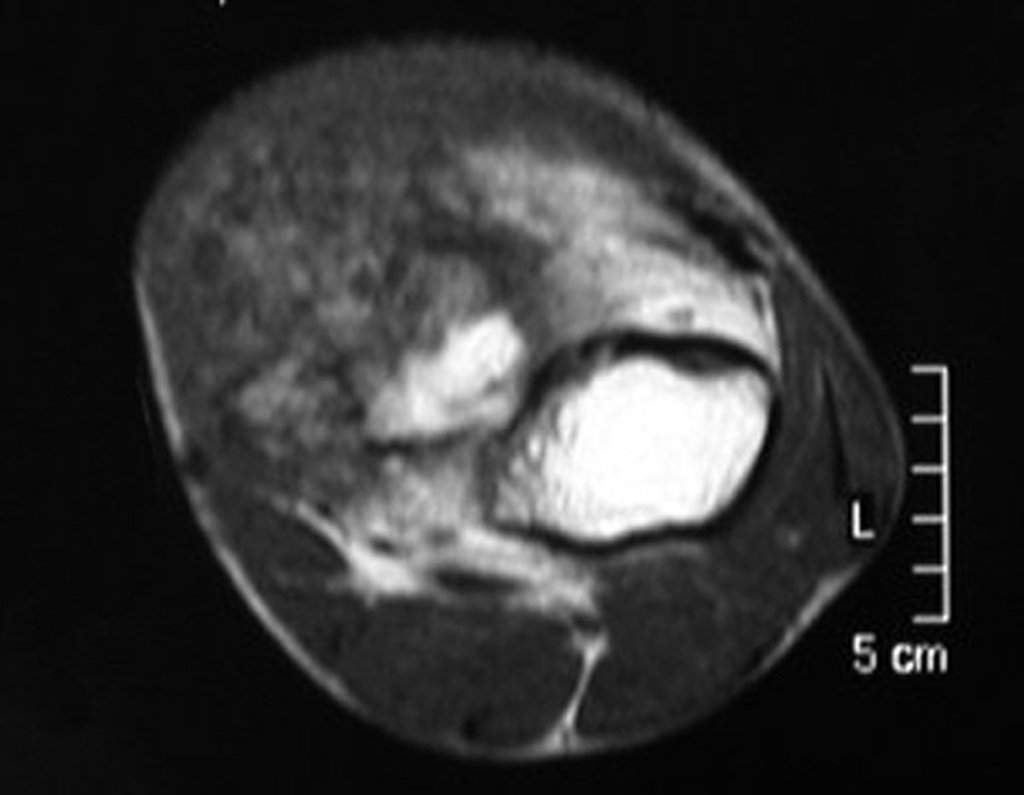
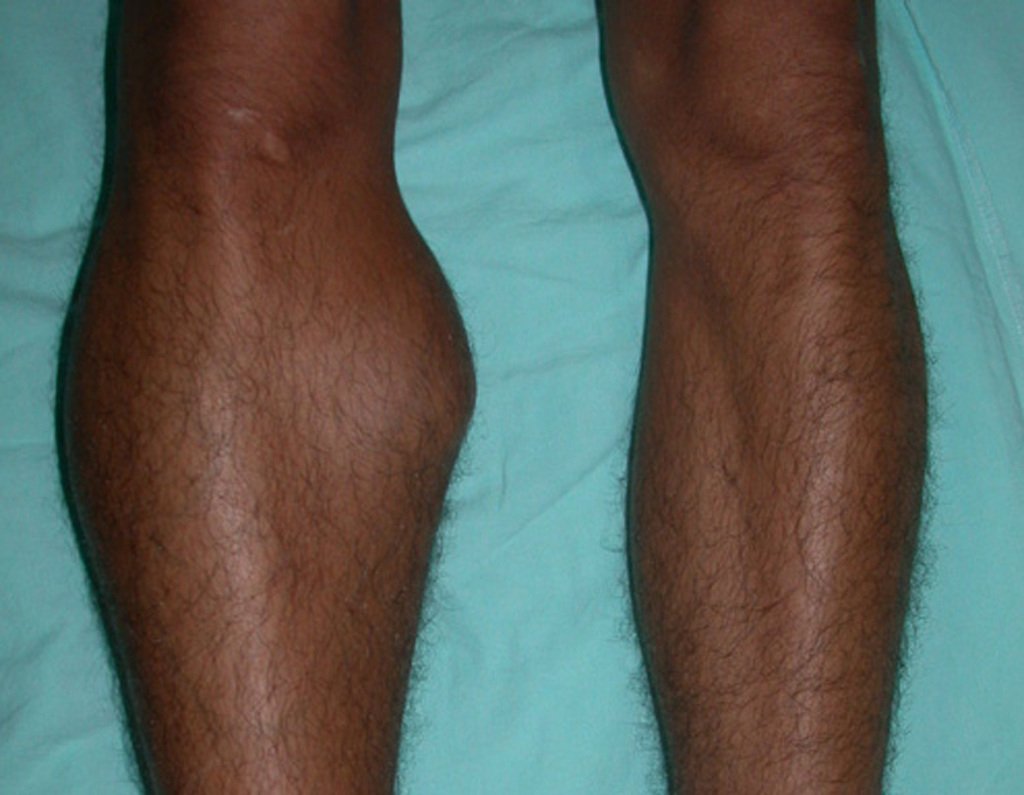
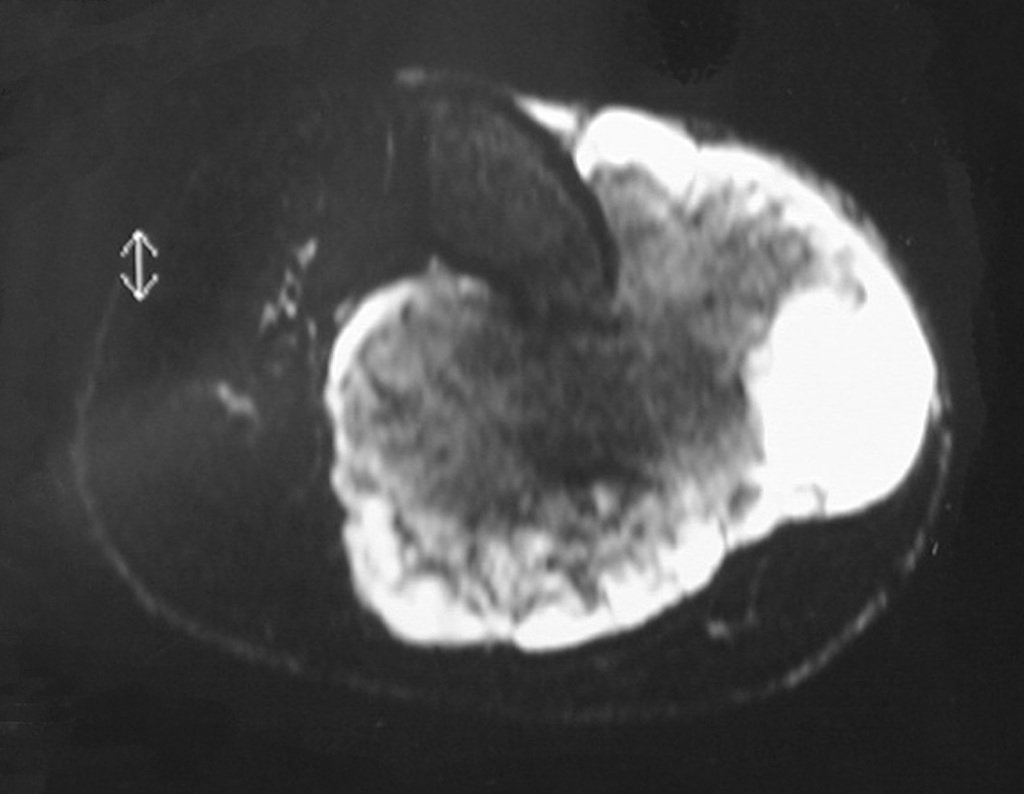
Differential diagnosis:
It presents a differential diagnosis with myositis ossificans, chondromyxoid fibroma, GCT, non-Hodgkin lymphoma 6,23,29 and aneurysmal bone cyst, due to its multiloculated nature. Histologically, the juxtacortical subtype resembles chondroma, osteochondroma, chondroblastoma and surface osteosarcoma 16 .
Clear cell chondrosarcoma has malignant chondrocytes with clear cytoplasm, osteoclast-like giant cells, and intralesional reactive bone formation causing confusion with osteosarcoma.
Mesenchymal chondrosarcoma is formed by islands of well-differentiated hyaline cartilage surrounded by sheets of small, round cells, reminiscent of hemangiopericytoma and Ewing’s sarcoma 14.
Central chondroma of long bones, chondrosarcoma and bone infarction are often difficult to differentiate, requiring clinical and radiographic monitoring to assess whether or not the lesion has progressed, before defining the course of action. Biopsy
is often not definitive for diagnosis 12,23,29.
Treatment:
The treatment of chondrosarcoma is surgical 25 , and a wide resection must be chosen, including the biopsy path 13,21.
Radiotherapy is ineffective 6 in controlling this neoplasm. For high-grade lesions, it is possible to discuss the indication of chemotherapy using the protocol for large cell sarcomas, based on anthracyclines 9999. For mesenchymal chondrosarcoma, which presents a predominance of small and undifferentiated cells, chemotherapy, when indicated, falls under the protocol of treatment of Ewing Tumor. 888
In both cases, the response to chemotherapy is usually poor 6 . The treatment of this neoplasm must be individualized for each clinical subtype:
– Central chondrosarcoma has high cure rates with appropriate surgery, therefore its treatment with intralesional curettage cannot be underestimated, even followed by complementary adjuvant methods, whether with phenol, liquid nitrogen, electrothermia or CO 2 laser 21.
Therefore, in cases of diagnostic doubt between chondroma and grade I chondrosarcoma, it is preferable to observe the evolution of this lesion, as it is known that the biopsy will not be conclusive, as the histological differential diagnosis between chondroma and grade I chondrosarcoma is difficult.
In some cases, these lesions can be treated with conservative surgery without performing a prior biopsy 21 .
When imaging tests: radiography, tomography and magnetic resonance, show a central lesion, without erosion of the internal cortex, with a casual and painless finding, it should be reevaluated initially within three months, if unchanged, repeated within six months and If the lesion remains unchanged, annual reassessments are scheduled.
If, at any time, there is a change in the clinical picture or imaging, it should be treated as central chondrosarcoma, carrying out wide resection of the lesion and reconstruction with non-conventional endoprosthesis, osteosynthesis with autologous or homologous graft or ablative surgery as necessary. of each case.
In the experience of these authors, it is unnecessary to operate on a painless chondroma when it is found casually, without radiographic aggressive characteristics. Performing an intralesional curettage, with local adjuvant and graft or cement, will not eliminate the need for careful observation. If the anatomopathological examination of the entire curettage reveals that it was chondrosarcoma, it will be much worse to re-operate on this region that has already been surgically manipulated.
There are several cases of “chondroma” in which the histology of intralesional curettage corroborated the biopsy appearance of “chondroma” and however had an unfavorable outcome. When monitoring these patients, imaging tests revealed that there was a “new” lesion at the site and that it was now chondrosarcoma.
In these curettages, local and distant dissemination and even dedifferentiation of the chondrosarcoma may occur, significantly worsening the prognosis.
– Juxtacortical chondrosarcoma, treatment is essentially surgical, with partial parietal resection EXAMPLE possible when possible, an effective procedure with lower morbidity compared to segmental resection.
– Peripheral chondrosarcoma , secondary to osteochondroma, care must be taken especially with the surface of the lesion, which presents anaplasia.
The surrounding soft tissue perimysium should be removed as an oncological margin to prevent local recurrence.
It is important to highlight that when there is growth of a bony exostosis after skeletal maturity, heterogeneous calcification, thick cartilaginous cap, unrelated to friction or trauma, it is probably a chondrosarcoma.
In this situation, a negative biopsy sample does not exclude the possibility of malignancy in the remainder of the lesion, and resection surgery with an oncological margin must be performed, paying special attention to the surface of the lesion.
– Mesenchymal chondrosarcoma , in addition to the need for local control with extensive surgery, may eventually be indicated for additional chemotherapy treatment 9999 .
– Dedifferentiated Chondrosarcoma , such as Clear Cell Chondrosarcoma, local control must be carried out with extensive surgery and chemotherapy with cisplatin and doxorubicin 9999.
Complications:
Intralesional curettage of chondrosarcoma can lead to local recurrence of the disease and more aggressive histological dedifferentiation.
In cases of dedifferentiated chondrosarcomas, hematogenous metastases to the lungs are frequent, which may present lymphatic dissemination and local recurrence 29 . Many chondrosarcomas tend to spread locally 14 , reaching enormous sizes and becoming inoperable, causing death due to compression or complications from this local spread.
Local recurrence increases the incidence of lung metastases 21.
Bibliography
1. ACKERMAN, L.V.; SPJUT, HJ Tumors of bone and cartilage. Atlas of tumor pathology. Washington, Air Force Inst. Pathology, 1962, fasc, 4.
2. CANALE, ST Campbell.Barueri orthopedic surgery: Manole; 2006
3. DAHLIN, DC Bone tumors. Barcelona: Ediciones Toray S/A; 1982
4. DORFMAN, HD; CZERNIAK, B. Bone tumors. St Louis, CV Mosby Co., 1997, chap. 7, p.410.
5. EDEIKEN, J.; HODES, PJ Radiological diagnosis of human illnesses. Buenos Aires, Panamericana, 1977, chap. 15.
6. ETCHEBEHERE, M. Malignant cartilaginous tumors: Chondrosarcomas. In: Camargo OP Clínica Ortopédica. Rio de Janeiro: Med si; 2002. p. 753-759
7. FELDMAN, F. Cartilaginous tumors and cartilage-forming tumor like conditions of the caps and soft tissues. In: Diseases of the Skeleton System (Roentgen Diagnosis). Part. 6 – Bone Tumors, New York, Springer-Verlag, 1977, p.177.
8. FLETCHER, CDM, Unni KK, WHO – Merters F. (Eds.): World Health Organization. Classification of Tumors. Pathology and Genetics of Tumors of Soft Tissue and Bone. IARC Press: Lyon 2002.
9. GREENSPAN, A. Orthopedic radiology. Rio de Janeiro: Guanabara; 2001.
10. HENDERSON, ED; Le PAGE, GA Apud FELDAMAN, F. Cartilaginius tumors and cartilage forming tumor like conditions of the bone and soft tissues. In: Disease of the Skeletal System (Roentgen Diagnosis).
Part. 6 – Bone tumors, New York, Springer Verlag, 1977, p.182.
11. HUVOS, AG Bone tumors Diagnosis, Treatment and Prognosis. Philadelphia, WB Saunders Co., 1979, p. 13.
12. JAFFE, HL Tumors and tumoral states of bones and joints. Mexico: La Prensa Medica Mexicana;1966.
13. JESUS-GARCIA, R. – Reynaldo Jesus-Garcia
14. LICHTENSTEIN, L. Barcelona: Talleres Graphics Ibero-Americanos; 1975.
15. LICHTESTEIN, L. Bone Tumor. 4 Ed St. Louis, CV Mosby Co., 1972, chap. 15.
16. LICHTESTEIN, L.; BERNSTEIN, D. Unusual benign and malignant chondroid tumors of bone. Cancer, 12:1142, 1959.
17. MARCOVE, RC Chondrosarcoma: Diagnosis and treatment. In: Orthopedic Clinics of North America. Tumors of the musculoskeletal apparatus. Buenos Aires, Panamericana, 1977, chap. 7.
18. MARCOVE, RC et al. Chondrosarcoma of the pelvis and upper end of the femur. In the analysis of factors influencing survival time in 113 cases. J. Bone Joint Surg., 54A:61, 1972.
19. MARCOVE, RC; SHOJI, H.; HARLEN, M. Altered carbohydrate metabolism in cartilaginous tumors. Contemp. Surg. 5:53, 1974.
20. McFARLAND, GBJr.; McKINLEY, L.M.; REED, RJ Dedifferentiation of low grade chondrosarcomas. Clinic. Orthop., 122:157, 1971.
21. MENENDEZ, LR Orthopedic knowledge update: Updates in orthopedic surgery and traumatology. Barcelona: Ars Medica; 2003.
22. O’NEAL, LW; ACKERMAN, LV Chondrosarcoma of cap. Cancer, 5:551, 1952.
23. PROSPERO, JD Bone Tumors. São Paulo, Roca, 2001, chap. II.
24. ROBBINS. Structural and functional pathology. Rio de Janeiro: Guanabara; 1996.
25. ROMSDAHL, M.; EVANS, H.L.; AYALA, AG Surgical treatment of chondrosarcoma. In: Management of primary bone and soft tissue tumors. Chicago, Year book med. Publisher Inc., 1977, p. 125.
26. ROMSDAHL, M.; Evans, H.L.; Ayala, AG Surgical treatment of chondrosarcoma. In: Management of primary bone and soft tissue tumors. Chicago. Year book med. Publisher Inc., 1977, p.125.
27. SAVIOR, AH; BEABOUT, JW; DAHLIN, DC Mesenchymal chondrosarcoma. Cancer, 28:605, 1971.
28. SCHAJOWICZ, F. Juxtacortical Chondrosarcoma. J. Bone Joint. Surg., 59B:473, 1977.
29. SCHAJOWICZ, F. Tumors y Lesiones Seudotumorales de Huesos y Articulaciones. Buenos Aires: Editora Médica Panamericana; 1982.
30. TORNBERG, DN; RICE, R.W.; JOHNSTON, AD The ultrastructure of chondromyxoid fibroma.Clin. Orthop. Rel. Research, 95:295, 1973.
999. J Clin Oncol 30:abstract 100:23,2012(maluf)
888. Buzaide, AC; Maluf, FC; Rocha Lima, CM
Brazilian Clinical Oncology Manual. Dendrix Edition and Design ltda. São Paulo (XI) Adult Bone Sarcomas, 560-79. 2013